



Sclérose latérale amyotrophique
A.M.I.S. des M.O.M :: OBJECTIFS ASSOCIATION DE PATIENTS SOLIDAIRES/ OBJECTIVES OF PATIENT SOLIDARITY ASSOCIATION :: Cliquer ici- pour accéder aux différents sujets-FORUMS-DIVERS - diffusions LIENS - AMIS D MOM - Click here to reach the various subjects - FORUMS-

Sclérose latérale amyotrophique
par AMIS4 Ven 23 Oct - 22:51
La Sclérose latérale amyotrophique aussi appelée en France maladie de Charcot du nom de celui qui la décrit pour la première fois en 1865, est une maladie neuro dégénérative d'étiologie inconnue touchant les motoneurones de la corne antérieure de la moelle épinière, du tronc cérébral et du cortex.
AMIS4- Invité
Centre de référence : PROTOCOLE pour S.L.A.
par Admin Lun 2 Nov - 21:15
Ma cousine Patou est atteinte de la S.L.A.
Le diagnostic a été établi en novembre 2007.
Elle avait vu un généraliste pour une sorte de fourmillement gênant à un de ses poignets.
Ex-sportive de haut niveau (natation), elle n'avait jamais eu de problème de santé. Dès le premier entretien avec ce médecin et après 3 questions bien ciblées, elle a été prise en charge pour une MLD avec diagnostic de SLA.
Ne sachant absolument rien de la maladie et de ce qui l'attendait, elle s'est rendue "innocemment" à un rendez-vous pris parce médecin pour elle à la Salpêtrière.
Ce rendez-vous a été un énorme coup de massue puisqu'on lui a froidement et sans précaution annoncé qu'elle allait décéder sous 36 mois.
Au début de l'année 2009, elle a été intégrée dans un protocole que je serais bien incapable de nommer mais ce que je peux avancer, c'est que la dégradation s'est montrée plus rapide. Je l'avais vue en mars, elle ne pouvait plus se servir d'une main et commençait à perdre l'équilibre même assise.Trois mois plus tard, son élocution est devenue beaucoup plus dure à gérer, sa deuxième main ne répondait presque plus, elle était en fauteuil et elle avait des épisodes de difficultés respiratoires.
Elle vient de passer quelques semaines dans un centre (Escalades dans les Pyrénées) où elle a été admirablement prise en charge pour ses soins respiratoires.
Je sais que l'inéluctable est à prévoir mais ne peut-on faire en sorte que dans les statistiques, on ne se limite plus aux cas en cours?
Je veux dire par là qu'un cas déclaré, sachant que la durée de vie est très limitée, ne rentre plus dans les chiffres une fois le décès arrivé et forcément, il y aura toujours le même nombre de cas, donc, peu susceptible de justifier un budget plus important au niveau de la recherche.
***************
Bonjour Adana,
j'ai lu ton message avec beaucoup d'intérêt.
Si j'ai bien compris ta cousine a bénéficié des protocoles du Dr PRADAT; qui ont démarré à l'hôpital de la Salpétrière P. à Paris en 2008
Ma sœur a tenté de les intégrer, mais trop tard
sa respiration (examen de 03/2008) était trop faible : inf. à 50
elle est décédée en 11/2008
j'ai passé beaucoup de temps avec elle.
J'ai cherché désespérément des soutiens.
Il existe une association qui peut t'apporter des renseignements :
Il s'agit de L'ARS
Il me semble que des recherches ont été diligentées -
appel d'offres auprès des chercheurs.
Voilà si cela peut te servir des coordonnés à retenir :
La Maison de l'ARS
Permanence et accueil de 14h à 17h au numéro vert :
0 800 600 106
(ou au 01 43 38 99 89)
Les permanences de Mme Dorgans, assistante sociale, en lien avec Maître Benjamin Pitcho, consultant juridique, se tiennent tous les vendredis de 14h à 17h.
75 avenue de la République, 75011 Paris
(métro rue St -Maur)
Tél : 01 43 38 99 89
Fax : 01 43 38 31 59
E-mail : ars@wanadoo.fr
Tu peux retrouver également toutes les actions de l'ARS en région sur le site internet : www.ars.asso.fr
Emmanuel Hirsch, Président de l'ARS.
Adana,
On est très démuni face à la souffrance de nos proches.
pourtant il est nécessaire de s'accrocher.
Peux tu me dire quel âge a ta cousine si ce n'est pas indiscret ?
A t-elle eu un choc qui aurait provoqué plus rapidement cette pathologie ?
Lui a t-on prodigué un traitement ? autre que ces cachets très onéreux qui retardent un peu.
Lui a t-on fait des Injections - bras ou autres ? (objet des protocoles)
a t-elle eu des Réactions ? ou améliorations.
Connait-elle la forme de sa pathologie ?
forme bulbaire ou pas ?
Je suis à ta disposition pour en discuter.
à bientôt

Admin- Admin
- Messages : 168
Date d'inscription : 15/10/2009 -

définitions S.L.A.
par AMIS4 Dim 29 Nov - 20:05
Maladie de Charcot
Maladie de Lou Gherig
La maladie
Le diagnostic
Les aspects génétiques
Le traitement, la prise en charge, la prévention
Vivre avec
En savoir plus
Madame, Monsieur,
Cette fiche est destinée à vous informer sur la sclérose latérale amyotrophique. Elle ne se substitue pas à une
consultation médicale. Elle a pour but de favoriser le dialogue avec votre médecin. N’hésitez pas à lui faire
préciser les points qui ne vous paraîtraient pas suffisamment clairs et à demander des informations supplémentaires
sur votre cas particulier. En effet, certaines informations contenues dans cette fiche peuvent ne pas être adaptées à votre cas : il faut se rappeler que chaque patient est particulier. Seul le médecin peut
donner une information individualisée et adaptée.
l A quoi est-elle due ? Comment expliquer les symptômes ?
La SLA est due à une dégénérescence progressive de certaines cellules nerveuses (ou neurones),
les motoneurones. Les motoneurones permettent de transmettre les ordres de mouvement
(envoyés par le cerveau) aux muscles qui vont effectuer le mouvement commandé
(figure 1). Ces neurones sont situés dans le cerveau (dans la zone appelée cortex moteur),
dans la moelle épinière, qui est une sorte de cordon situé à l’intérieur de la colonne vertébrale,
prolongeant le cerveau, ainsi que dans le bulbe rachidien (qui assure la jonction
entre le cerveau et la moelle épinière).
Au cours de la SLA, les motoneurones meurent (ou dégénèrent) progressivement. Par conséquent, les muscles qui ne sont plus stimulés deviennent inactifs, s’affaiblissent et perdent du volume. On parle d’atrophie musculaire ou d’amyotrophie.
A mesure que la maladie évolue, les activités simples comme le langage, la marche, les mouvements
des mains, la déglutition, deviennent progressivement difficiles ou impossibles.
On ne connaît pas les causes exactes de la dégénérescence des motoneurones, mais plusieurs théories sont actuellement discutées par les scientifiques. Il a notamment été mis en évidence que des personnes atteintes de SLA présentaient des anomalies au niveau du glutamate, un « messager » qui intervient dans la transmission des messages nerveux. Un niveau trop élevé de glutamate pourrait être responsable de l’épuisement des neurones.
Une autre hypothèse évoque un dérèglement d’un « facteur de croissance »,
Les motoneurones prennent le relais, dans la moelle épinière, du message envoyé par le cerveau par un premier
neurone (neurone pyramidal) pour bouger un muscle. Les motoneurones sont reliés directement aux muscles, par leurs
prolongements qui constituent les nerfs. Il existe aussi des motoneurones situés dans le cerveau.
La sclérose latérale amyotrophique
permet de favoriser la croissance de certaines cellules, qui pourrait entraîner la dégénérescence
des motoneurones.
Il semble également qu’une réaction d’inflammation anormale puisse survenir au niveau des motoneurones. Une autre piste concerne les éléments qui produisent l’énergie au sein des cellules, les mitochondries. Dans les motoneurones lésés dans la SLA, les mitochondries semblent en voie de dégénérescence, ce qui pourrait expliquer la mort des motoneurones.
Enfin, chez les personnes atteintes de SLA, les mécanismes naturels de mort cellulaire (apoptose) pourraient se déclencher trop tôt et trop rapidement.
La SLA pourrait être due à une combinaison de ces différentes hypothèses, qui font toutes l’objet de programmes de recherche.
Est-elle contagieuse ?
La SLA n’est pas contagieuse.
Quelles en sont les manifestations ?
Tous les muscles qui assurent les mouvements du corps (muscles moteurs) peuvent être touchés par la maladie, notamment les muscles des bras, des jambes, de la parole, de la déglutition et de la respiration. Cependant, quelle que soit la forme que prenne la maladie, les fonctions sensorielles (capacité de goûter, voir, sentir, entendre et toucher), les fonctions sexuelles, urinaires et les fonctions intellectuelles du malade ne sont pas altérées.
La dégénérescence des neurones n’entraîne pas de douleurs « physiques » à proprement parler, mais certaines douleurs musculaires ou articulaires sont souvent rencontrées.
La SLA peut se présenter sous deux formes principales :
la forme « spinale » (qui débute par l’atteinte d’un membre), et la forme « bulbaire » (qui débute par l’atteinte des muscles de la bouche). La forme spinale représente deux tiers des cas, et affecte davantage les hommes vers l’âge de 55 ans. Elle est due à la dégénérescence des motoneurones situés dans la moelle épinière.
La forme bulbaire concerne plutôt les femmes et apparaît vers 60-65 ans, et correspond à la dégénérescence des motoneurones situés dans la région du cerveau qui commande, entre autres, les mouvements de la langue et du palais (le « bulbe rachidien »).
Ces deux formes peuvent se succéder ou se développer simultanément, la maladie progresse presque toujours vers une forme complète (spinale et bulbaire).
Forme spinale
La maladie débute en général par une sensation de faiblesse d’une partie d’un membre, accompagnée de petites contractions ou secousses musculaires involontaires (fasciculations).
Ces contractions s’accompagnent de crampes, de contractures douloureuses et d’une sensation de raideur (ou ankylose) dans les articulations et les membres, et rendent peu à peu les mouvements difficiles. Les premiers symptômes incluent des difficultés de coordination des mouvements, le manque de précision de certains gestes (ou le fait de lâcher involontairement un objet), une gêne à la marche, des troubles de l’équilibre, et/ou des chutes.
Ces troubles s’accentuent peu à peu et s’associent à une fatigue générale. Tous les membres finissent par être atteints, mais de façon asymétrique (un côté est plus touché que l’autre).
La fonte musculaire (amyotrophie) survient plus ou moins rapidement. Le malade maigrit, souvent en partie à cause de l’amyotrophie.
Forme bulbaire
Dans la forme bulbaire, les premiers symptômes sont des difficultés à articuler ou à prononcer certains mots, des changements de voix (elle devient rauque, faible ou nasillarde).
Le malade éprouve également des difficultés à mâcher, à bouger la langue et le visage, et à
avaler (et donc à manger et à boire). Même le fait d’avaler sa salive devient source de problème : le malade avale souvent « de travers » (on parle de fausses routes, qui correspondent au passage de la salive ou des aliments dans les voies respiratoires).
L’affaiblissement des muscles de la bouche et de la gorge conduit en effet à des problèmes de déglutition,
elle ne se fait plus automatiquement. Le malade est souvent gêné par une salivation abondante (hypersalivation) ou au contraire par une sécheresse excessive de la bouche.
Certaines personnes atteintes subissent des périodes de rires ou de pleurs involontaires, ce qui est très décontenançant pour l’entourage.
L’évolution se fait vers une aggravation progressive, sur des années. Une fatigue importante, souvent matinale, accompagne les autres symptômes.
Dans les stades plus avancés de la maladie, des difficultés à respirer surviennent parce que les neurones qui contrôlent les muscles respiratoires sont atteints.
D’autres symptômes apparaissent, comme une constipation, un amaigrissement important, des troubles de la circulation sanguine dus à l’immobilité (avec parfois des sensations de picotements sur la peau, les paresthésies), et des troubles du sommeil.
Les capacités intellectuelles ne sont pratiquement jamais altérées par la SLA : le malade reste donc tout à fait lucide tout au long de la maladie.
Quelle est son évolution?
La SLA est une maladie neurodégénérative, ce qui signifie que le processus de destruction des neurones se poursuit tout au long de la maladie et est inévitable. Il s’agit donc d’une maladie qui devient rapidement handicapante (au niveau moteur) et qui réduit considérablement l’espérance de vie. La maladie évolue à un rythme différent d’une personne atteinte à l’autre, et il est impossible de prévoir la durée d’évolution de la maladie.
La forme de SLA à début bulbaire se caractérise par une évolution plus rapide. L’espérance de vie d’une
personne atteinte de la SLA est d’environ 3 à 5 ans après le diagnostic. Cependant, avec l’amélioration de la prise en charge, 20 % des personnes atteintes vivent cinq ans ou plus après le diagnostic, et 10 % vivent plus de 10 ans ou plus.
Il existe aussi des formes bénignes de la maladie qui restent stables sur plus de 30 ans, mais elles sont rares.
Les difficultés à respirer liées à la paralysie des muscles respiratoires et aux infections respiratoires (qui peuvent être favorisées par les troubles de la déglutition) sont la cause la plus fréquente de décès.
Le diagnostic
Comment fait-on le diagnostic de cette maladie ?
Quels sont les examens complémentaires ? A quoi vont-ils servir ?
Il n’existe pas de test spécifique pour diagnostiquer une SLA. Comme les premiers symptômes
peuvent être assez discrets (crampes, faiblesse de la main, modification de la voix).
les médecins ont parfois du mal à faire le diagnostic. Ils doivent avant tout « éliminer » les maladies proches de la SLA.
Pour ce faire, un électromyogramme (EMG) est systématiquement effectué. Il s’agit d’un examen qui permet d’évaluer l’atteinte des muscles à l’aide de petites aiguilles (électrodes) mises en contact avec les différents muscles. En cas de SLA, l’électromyogramme révèle que les muscles fonctionnent bien, mais que les nerfs (et donc les motoneurones) qui leur donnent les ordres sont affaiblis. On parle de « dénervation », qui survient avant tout au niveau des muscles des membres, du cou, et du thorax.
Le scanner ou l’IRM (imagerie par résonance magnétique) sont également utilisés pour vérifier que les symptômes ne sont pas dus à une blessure ou une anomalie de la moelle épinière (située dans la colonne vertébrale) ou du cerveau.
Une ponction lombaire peut être réalisée. Elle consiste à prélever le liquide circulant autour de la moelle épinière (le liquide céphalo-rachidien) pour vérifier qu’il n’y a pas d’infection.
La ponction lombaire se fait à l’aide d’une aiguille enfoncée à l’intérieur de la colonne vertébrale dans le bas du dos.
L’analyse du liquide est normale en cas de SLA.
Peut-on confondre cette maladie avec d’autres ? Lesquelles ?
Comment faire la différence ?
Un certain nombre d’affections proches de la SLA se manifestent aussi par des faiblesses musculaires, des crampes, des petites contractions musculaires… et notamment :
- les tumeurs situées aux alentours de la moelle épinière et provoquant sa compression, et donc des problèmes de transmission des messages nerveux
- certaines intoxications (notamment au plomb, à l’arsenic et à l’aluminium)
- certaines maladies infectieuses à manifestations neurologiques, dont la brucellose, la maladie de Lyme…
- les maladies de la moelle épinière (myélopathies) ou certaines maladies musculaires
- les autres maladies des neurones moteurs ou des nerfs, et notamment les amyotrophies spinales, la maladie de Kennedy, la neuropathie motrice multifocale avec blocs de conduction…
L’IRM de la moelle épinière et du cerveau et l’examen du liquide céphalo-rachidien permettent d’écarter certaines de ces affections.
Peut-on dépister cette maladie avant qu’elle ne se déclare ?
Non, on ne peut pas dépister cette maladie avant qu’elle ne se déclare.
Les aspects génétiques
Quels sont les risques de transmission ?
Dans la majorité des cas, la maladie est sporadique, c’est-à-dire qu’elle atteint une personne de manière isolée.
Cependant, dans 5 à 10 % des cas, la SLA est « familiale » (elle touche plusieurs membres d’une même famille).
Les formes familiales se déclarent souvent plus tôt (avant 50 ans en moyenne)
Et leur évolution est généralement plus rapide, mais les symptômes sont les mêmes que ceux des
SLA non familiales.
On sait que 10 à 20 % des formes familiales de SLA sont dues à l’anomalie (mutation) d’un
gène appelé SOD1. Pour les 80 à 90 % des SLA familiales restantes, d’autres gènes sont
impliqués mais ne sont pas encore clairement identifiés.
Si c’est le gène SOD1 qui est en cause, la transmission est autosomique dominante, ce qui
signifie que la maladie peut se transmettre de génération en génération, un malade ayant
un risque sur deux de transmettre la maladie à chacun de ses enfants à chaque grossesse,
quel que soit leur sexe. Cela concerne environ 2 % de toutes les SLA.
Quels sont les risques pour les autres membres de la famille ? Peut-on faire un dépistage ?
Le risque, pour un autre membre de la famille, de développer une SLA, est faible, sauf dans
les formes familiales.
A l’heure actuelle, en l’absence de connaissances de tous les gènes impliqués dans les formes familiales, il n’est pas possible de déterminer avec certitude si une personne malade est, ou non, atteinte d’une forme familiale par un examen biologique. La seule méthode disponible est l’investigation de l’histoire de la famille, à la recherche d’autres cas, suivie d’un examen génétique à la recherche d’une mutation présente chez les personnes malades
et absente chez les personnes non malades de la famille (seule façon de prouver qu’il y a un lien entre la mutation et la maladie, car beaucoup de mutations n’entraîne pas de maladie).
Le traitement, la prise en charge, la prévention
Existe-t-il un traitement pour la SLA ?
Il n’existe pas de traitement qui guérit cette maladie. Un seul médicament, le riluzole, permet de ralentir l’évolution de la maladie. Il diminue le taux de glutamate, ce messager nerveux qui se trouve peut-être en trop grande quantité chez les personnes atteintes de SLA. Il est en général prescrit dès que la maladie est suspectée.
En outre, certains médicaments permettent d’atténuer les symptômes (voir « quelles sont les autres options thérapeutiques ? ») et des mesures non médicamenteuses peuvent être mises en place pour soulager et accompagner les personnes atteintes. Ainsi, un travail de maîtrise de soi et de ses émotions, un accompagnement psychologique (voir « Un soutien psychologique est-il souhaitable ? »), des séances de kinésithérapie, de rééducation ou d’orthophonie peuvent aider les malades à maintenir la souplesse des muscles, et à conserver leur autonomie et leur capacité à communiquer le plus longtemps possible.
Marche et mouvements
L’affaiblissement musculaire progressif entraîne des chutes imprévisibles souvent dangereuses (blessures à la tête, fractures…), surtout qu’au début de la maladie, le désir de continuer à faire des activités (marcher, faire du vélo) est tel qu’il peut conduire à « ignorer » les symptômes. Pour éviter de se blesser, ce qui risquerait d’accélérer l’évolution de la maladie, il est donc recommandé de s’aider d’une canne si besoin. Les séances de kinésithérapie sont primordiales pour maintenir la souplesse des muscles. Cependant, les exercices ne permettront aucunement de redonner de la force aux muscles affaiblis : ils permettent simplement d’entretenir les fonctions musculaires restantes.
Des pratiques douces, à faire soi-même, éventuellement accompagnées de techniques de relaxation (respiration, concentration, massages, étirements…) peuvent également aider à maintenir sa capacité musculaire le plus longtemps possible.
Lorsque la marche devient de plus en plus pénible et risquée, des aides techniques peuvent être proposées au malade : un déambulateur, des orthèses (appareillages destinés à soutenir un membre ou une articulation), puis un fauteuil roulant. Accepter de se servir d’un fauteuil roulant est une étape psychologique difficile à franchir, mais cela peut permettre « d’économiser » de l’énergie pour d’autres activités et de rester ainsi plus autonome.
Aménagement du domicile
Un ergothérapeute, personne qui aide à la rééducation des personnes atteintes de déficience physique ou psychique, peut évaluer les difficultés qu’a le malade à accomplir des gestes simples : se lever de son lit, s’asseoir, prendre sa douche… Il pourra mettre en oeuvre des aides matérielles (barres d’appui par exemple), techniques (apprendre à faire ces gestes autrement), et éventuellement suggérer l’intervention d’une aide à domicile.
L’aménagement du domicile est souvent nécessaire pour assurer le plus de confort possible au malade, surtout s’il se déplace en fauteuil roulant. L’installation d’un lit ou d’un matelas spécialement conçu pour les personnes handicapées peut apporter un réel confort au malade.
Communication
Au même titre que les séances de kinésithérapie, une rééducation du langage et de la déglutition auprès d’un orthophoniste permet de préserver au mieux l’usage de la parole mais aussi de limiter les troubles de la déglutition et donc de rendre les repas plus sûrs (en diminuant le nombre de fausses routes). Lorsque la parole ou le mouvement des mains (tourner les pages d’un livre par exemple) deviennent impossibles, plusieurs moyens de communication et de distraction mis au point pour les personnes handicapées permettent de maintenir le contact avec son entourage, de naviguer sur Internet, de lire…
Ainsi, des tourne-page électriques et une grande variété d’appareils d’aide à la communication, de synthètiseur de la parole à partir d’un texte, des logiciels de composition de phrases assistée, offrent un peu d’autonomie aux malades paralysés.
Il existe des ordinateurs configurés pour ces malades, avec des contacteurs (sortes de souris) adaptés aux capacités de chacun (c’est-à-dire actionnés par l’endroit ou la fonction du corps qui peut encore être mobilisé : souffle, joue, doigt…).
Alimentation
L’amaigrissement est généralement très important au cours de la SLA. Il est dû à la fonte musculaire, mais aussi aux difficultés croissantes d’alimentation, à l’inconfort ressenti lors des repas, mais aussi parfois à une perte d’appétit liée à la dépression. De nombreux malades sont en état de dénutrition ou de déshydratation, ce qui ne fait qu’aggraver leur faiblesse musculaire.
Une consultation auprès d’un diététicien peut permettre de mettre en place un régime adapté aux besoins nutritionnels du malade, en y associant éventuellement des compléments alimentaires. Pour limiter les troubles de la déglutition et donc l’angoisse d’une fausse route et d’un étouffement, les repas peuvent être mixés et l’eau peut être épaissie avec un gélifiant pour être plus facile à avaler. Certaines techniques, enseignées par l’équipe soignante, peuvent aider à limiter les fausses routes (positionnement adapté de la tête, de la cuillère, de la tasse…).
Cependant, au fur et à mesure de l’évolution de la maladie, les personnes atteintes auront de plus en plus de mal à prendre leurs repas « normalement ». Une aide à l’alimentation devient alors nécessaire et une gastrostomie peut être mise en place. La gastrostomie est une intervention qui consiste à poser un petit tuyau plastique reliant directement l’estomac à la paroi extérieure du ventre. Elle a pour but de mettre en place une sonde qui permettra
d’introduire des aliments liquides directement dans l’estomac (nutrition entérale). La gastrostomie n’empêche pas de maintenir en parallèle une alimentation « normale » bien qu’en moindre quantité, afin de préserver le plaisir de manger certaines choses.
Ces mesures invasives permettent de s’alimenter mieux et sans risque et offrent des bénéfices immédiats. Elles permettent de limiter les fausses routes et de diminuer ainsi le risque d’infection respiratoire. Il est recommandé d’installer la gastrostomie relativement tôt, pour que le malade puisse s’y habituer progressivement.
Respiration
La fonction respiratoire doit faire l’objet d’une attention et d’une surveillance particulières.
Lorsque les muscles respiratoires sont atteints, des séances de kinésithérapie respiratoire destinées à « évacuer » les sécrétions bronchiques doivent être mises en place. Elles sont réalisées au début par un kinésithérapeute mais l’entourage du malade peut apprendre à les pratiquer. En fait, elles permettent de faire tousser et expectorer le malade, parce que le mécanisme naturel de la toux ne fonctionne plus. Parfois, des appareils « d’aide à la toux »
(sous forme de masque naso-buccal) sont utilisés.
Cependant, la respiration des personnes atteintes de SLA finit le plus souvent par devenir insuffisante, créant des troubles du sommeil, des maux de tête, un essoufflement, une somnolence, une fatigue… Le malade (et sa famille) doit alors envisager l’emploi ou non d’un système de ventilation mécanique (oxygénation artificielle).
Une ventilation non invasive (VNI) est utilisée en premier lieu. Elle fait appel à un appareil qui envoie de l’air par l’intermédiaire d’embouts placés dans les narines, ou par un masque nasal ou naso-buccal (posé sur le nez et la bouche).
En fonction des difficultés respiratoires, la durée de ventilation peut être en premier lieu partielle (la nuit, quelques heures dans la journée), puis totale (24 heures sur 24).
En cas d’infection pulmonaire qui aggrave les difficultés respiratoires, un tube glissé par le nez dans la trachée (intubation) et relié à un respirateur peut être utilisé de façon provisoire en attendant la guérison de l’infection.
Lorsque les différents modes de ventilation non invasive ne sont plus assez efficaces ou lorsque les séances de ventilation sont de plus en plus longues, il est important d’aborder le sujet de la trachéotomie, qui peut ou non être effectuée. Une trachéotomie est une ouverture faite dans la trachée par chirurgie, au niveau du cou, qui permet de faire passer un tube (canule) qui, relié au respirateur, assurera l’assistance respiratoire (figure 2).
La mise en place d’une trachéotomie doit relever, dans la mesure du possible, d’une discussion entre l’équipe médicale et la personne concernée et son entourage. Ces derniers, pour évaluer les avantages et les inconvénients de ce geste, doivent bénéficier au préalable d’une information claire et complète. Il s’agit en effet d’une ventilation « invasive » définitive, qui marque une étape de la maladie et prolonge la durée de vie des personnes atteintes. Il est donc important que chaque malade puisse rester maître des décisions qui concernent son maintien en vie. Or, la trachéotomie est souvent réalisée dans l’urgence par une équipe médicale appelée en cas de « détresse » respiratoire du malade. Avant qu’une intervention d’urgence soit nécessaire, il est donc préférable que le malade ait exprimé
clairement son désir ou non d’être maintenu en vie de manière artificielle.
Quelles sont les autres options thérapeutiques ?
En plus de ces mesures d’aide à la vie quotidienne, les principaux symptômes associés à la SLA (crampes, constipation, troubles du sommeil, dépression…) peuvent faire l’objet d’un traitement par médicaments.
Crampes et douleurs
Les crampes musculaires sont très courantes chez les personnes atteintes de SLA. Il est important de garder les muscles au chaud, et de les étirer doucement ou les faire étirer par un kinésithérapeute. Des médicaments comme la quinine sont alors recommandés en plus. D’autres médicaments qui permet de décontracter les muscles myorelaxants, de type baclofène ou dantrolène) sont parfois utiles.
Les douleurs musculaires ou articulaires qui surviennent en raison de la fonte des muscles peuvent être soulagées par des étirements après chaque contraction et, si cela ne suffit pas, par des anti-douleurs (antalgiques) forts paracétamol et codéine, morphine).
Fatigue
La fatigue, surtout matinale, est l’un des symptômes principaux de la SLA. Il est recommandé de faire des siestes régulières et de limiter les activités qui aggraveraient la fatigue.
Les médicaments stimulants sont déconseillés.
Figure 2
Trachéotomie
1 – cordes vocales et trachée
2 - cartilage thyroïde
3 - cartilage cricoïde
4 – cartilages de la trachée
5 – ballon de fixation de la canule (tube)
Anxiété et dépression
Ce sont des symptômes très fréquents au cours de la SLA. Des antidépresseurs tels que l’amitriptyline peuvent aider ponctuellement les personnes qui en ressentent le besoin.
L’équilibre psychologique peut être retrouvé et entretenu par un travail personnel ou de groupe encadré par un psychothérapeute.
Troubles du sommeil
Les troubles du sommeil sont dus entre autres à l’inconfort lié à l’absence de changement de position pendant la nuit. L’anxiété peut également être source d’insomnie. Dans certains cas, des médicaments comme les benzodiazépines, aux propriétés relaxantes et anxiolytiques, peuvent être recommandés. Cependant, en cas de difficultés respiratoires nocturnes, ces médicaments ne sont pas prescrits. Les exercices de relaxation et de respiration sont souvent d’une grande aide pour favoriser le sommeil.
Constipation
La constipation est un symptôme presque constant au cours de la SLA. Un régime approprié, certains massages abdominaux et des médicaments laxatifs comme le lactulose ou le sorbitol peuvent faciliter le transit intestinal.
Rires et pleurs incontrôlables
Les épisodes de rires ou de pleurs sans rapport avec l’état émotionnel sont très perturbants pour le malade et l’entourage. Certains médicaments, comme l’amitriptyline, un antidépresseur, peuvent réguler ces « crises ».
Hypersalivation et hygiène buccale
La salivation excessive peut poser des problèmes d’inconfort et d’image de soi. Des médicaments tels que l’amitryptiline ou la clomipramine, l’atropine, ou l’ipratropium bromure en spray peuvent être utilisés pour assécher la bouche ou épaissir la salive, mais sont la plupart du temps insuffisants.
Il devient difficile de se brosser les dents et donc de garder une bonne hygiène dentaire.
Les soins dentaires doivent être effectués par un dentiste qui connaît la maladie et ses effets. De plus, certains malades atteints de la forme bulbaire de la SLA souffrent d’un épaississement de la langue. Celle-ci peut être nettoyée à l’aide d’une petite éponge ou avec une serviette mouillée.
Un aspirateur de sécrétions peut éventuellement être employé pour nettoyer les résidus d’aliments et éliminer les excès de salive.
Infections respiratoires
La faiblesse des muscles respiratoires et les fausses routes alimentaires à répétition peuvent augmenter le risque d’infections pulmonaires. Des antibiotiques sont parfois prescrits en prévention.
Soins palliatifs
En phase terminale de la maladie, des soins palliatifs sont mis en place. Le but des soins
palliatifs est d’offrir la meilleure qualité de vie possible aux malades et de les accompagner
en fin de vie. Ce sont des traitements et des soins d’accompagnement physiques et psychologiques, prodigués en institution ou à domicile.
Quelles seront les conséquences du traitement pour la vie quotidienne ?
La vie quotidienne sera bouleversée par la maladie, et certains soins lourds devront éventuellement être prodigués par des aides à domicile, notamment la toilette, l’entretien des appareils de ventilation, les déplacements…
Si une trachéotomie est réalisée, l’adaptation du malade et de son entourage peut nécessiter du temps. Par ailleurs, des règles d’hygiène strictes doivent être respectées, par exemple pour aspirer les sécrétions (ou mucosités) qui se forment dans la trachée du malade. Une garde est indispensable en permanence, de jour comme de nuit, pour réaliser les aspirations.
Elle est la plupart du temps laissée à la charge des familles, ce qui nécessite une réorganisation complète du mode de vie.
Quels bénéfices attendre des traitements ?
Dans l’état actuel des connaissances et de la recherche, l’évolution de la SLA est telle que la dégénérescence des neurones est inévitable, quelles que soient les mesures entreprises pour diminuer les symptômes. L’encadrement et le soutien au malade, notamment par les thérapies physiques, permettent d’utiliser au mieux les capacités physiques de la personne malade l’autonomie le plus longtemps possible.
Les différents médicaments et un travail sur soi peuvent rendre certains aspects de la maladie
moins difficiles à vivre.
Quels sont les risques des traitements ?
Le riluzole est en général bien toléré. Il peut cependant entraîner des effets indésirables :
troubles digestifs, douleurs abdominales, étourdissements, accélération du rythme cardiaque (tachycardie), fatigue importante. Rarement, il entraîne des réactions allergiques qui nécessitent l’arrêt du traitement. Les autres médicaments proposés peuvent également avoir des effets secondaires gênants qu’il faut signaler au médecin.
Un soutien psychologique est-il souhaitable ?
A chaque stade de la maladie, un soutien psychologique est nécessaire pour le malade et pour son entourage.
L’annonce du diagnostic, et donc de la sévérité du pronostic, est un moment extrêmement difficile où se mêlent sentiments d’impuissance, de colère, d’injustice et de désespoir.
L’acceptation de la maladie peut prendre beaucoup de temps, et est d’autant plus difficile que l’évolution peut être extrêmement rapide et que le malade se sent vite prisonnier de son propre corps. Pour le malade, la perte progressive de son autonomie physique est d’autant plus pénible que ses facultés intellectuelles restent intactes et qu’il est parfaitement lucide, témoin de l’évolution de sa maladie. Au-delà de la perte de la mobilité, la perte de la parole et de la capacité à communiquer est particulièrement mal vécue. La SLA s’accompagne très fréquemment de symptômes dépressifs et d’un repli sur soi auquel un psychologue peut aider à faire face.
Le soutien psychologique est également important pour soutenir le malade dans ses décisions,
qui sont parfois contraires à ce que souhaiterait la famille, et pour l’aider à voir « clair » en mettant de côté son ressentiment et sa colère.
Par ailleurs, la maladie bouleverse l’entourage (conjoint et enfants surtout), et tous les médecins s’accordent à dire que les troubles psychologiques sont souvent plus importants chez ces derniers que chez les malades eux-mêmes.
Les « aidants » doivent donc bénéficier d’une aide psychologique qui permet d’améliorer leur qualité de vie mais également celle du malade. Cette aide permet aussi d’accompagner le malade dans toutes les démarches et
les décisions difficiles à prendre, en respectant ses choix parfois malgré soi.
Que peut-on faire soi-même pour se soigner ?
La participation active du malade (et de son entourage) aux soins et aux exercices de rééducation est primordiale pour l’efficacité et le bon déroulement de la prise en charge.
Comment se faire suivre ?
La prise en charge doit se faire dans un centre spécialisé dans la SLA.
L’état de santé des personnes atteintes y sera régulièrement évalué (bilan tous les 3 mois),
et notamment leur état nutritionnel et leur capacité respiratoire, pour éventuellement
mettre en place les mesures nécessaires.
Une évaluation de la force musculaire est également réalisée pour adapter le traitement et les appareillages.
S’il existe des troubles marqués du sommeil, un enregistrement du sommeil et de la respiration nocturne peut être réalisé chez soi ou à l’hôpital (oxymétrie nocturne).
Quelles sont les informations à connaître et à faire connaître en cas d’urgence ?
Il faut signaler le traitement en cours, pour éviter les interactions médicamenteuses, et faire part du diagnostic aux médecins. Si une décision concernant le désir ou non de réanimation a été prise par le malade, les proches doivent en faire part aux médecins urgentistes.
Par ailleurs, il existe une carte de soins et d’urgence établie par la Direction Générale de la Santé que le malade peut choisir de porter sur lui en permanence et qui donne aux médecins les instructions à suivre en cas d’urgence.
l Peut-on prévenir cette maladie ?
Non, on ne peut pas prévenir cette maladie aujourd’hui. On ne peut que ralentir son évolution.
Vivre avec
Quelles sont les conséquences de la maladie sur la vie familiale, professionnelle, sociale, scolaire ou sportive ?
Être atteint d’une maladie neurodégénérative est extrêmement difficile à vivre. La perte d’autonomie, parfois rapide, s’accompagne de la sensation pénible d’être une charge pour l’entourage.
Pour les proches, le sentiment d’impuissance et de détresse est également pesant. De plus, la maladie demande beaucoup d’énergie, de temps et nécessite une adaptation permanente aux besoins du malade (y compris au niveau social ou administratif)… Cependant, il ne faut pas perdre de vue qu’il y a une vie après le diagnostic et que la personne atteinte a besoin de « rebondir » pour accepter de vivre avec sa maladie et ses handicaps. Un accompagnement psychologique et un travail global de maîtrise de soi et de relaxation sont essentiels pour aider le malade à vivre avec la maladie. La prise en charge physique est également capitale car elle permet de maintenir une qualité de vie « acceptable » en prévenant les complications, en aidant à conserver une respiration efficace et surtout en préservant la communication. Les sorties (musées, restaurants, cinéma) et les déplacements sont possibles
à l’aide de quelques aménagements et une bonne organisation.
Le soutien de l’entourage est également primordial pour rassurer le malade et l’accompagner le mieux possible. Le soutien psychologique est important à toutes les étapes de la maladie, tant pour le malade que pour les aidants qui doivent aussi s’octroyer des périodes de « répit » pour se soulager. Cela étant, il existe malheureusement peu de structure d’accueil spécialisées pour les malades atteints de SLA.
L’aménagement des conditions de vie ou d’emploi nécessitent parfois l’intervention d’une assistante sociale et/ou d’un ergothérapeute.
De manière générale, les mesures palliatives doivent être évoquées posément et examinées en détail par la famille, les soignants et le malade qui doit mesurer toutes les conséquences de son choix. Il est préférable de prendre une décision dès que possible, avant qu’une intervention d’urgence soit nécessaire et qu’une réponse doive être donnée précipitamment, pour que les décisions de la personne atteinte puissent être respectées.
En savoir plus
Où en est la recherche ?
L’objectif principal de la recherche est de comprendre ce qui déclenche la maladie et quel est le mécanisme de dégradation des neurones. L’élaboration d’un traitement reste difficile compte tenu de l’état actuel des connaissances. Plusieurs essais cliniques sont néanmoins en cours pour évaluer l’efficacité de molécules qui pourraient ralentir l’évolution de la maladie.
Par ailleurs, les chercheurs étudient les formes familiales de SLA pour comprendre les causes de la maladie. Des souris qui possédent un gène SOD1 déficient, et qui présentent certains symptômes de la SLA, sont actuellement à l’étude.
Comment entrer en relation avec d’autres malades atteints
de la même maladie ?
En contactant les associations de malades consacrées à cette maladie. Vous trouverez leurs
coordonnées en appelant Maladies Rares Info Services au 0 810 63 19 20 (Numéro azur,
prix d’un appel local) ou sur le site Orphanet (www.orphanet.fr).
Renseignements :
Les prestations sociales en France
Il est important de trouver les bons interlocuteurs pour se faire aider dans les démarches administratives. Des conseils précieux peuvent notamment être fournis par les associations de malades qui sont au courant de la législation et des droits.
En France, les personnes qui souffrent d’une SLA peuvent bénéficier d’une prise en charge à 100 % par la Sécurité Sociale en ce qui concerne le remboursement des frais médicaux.
Le dossier de demande de reconnaissance d’affection longue durée (« ALD », qui impliquent une prise en charge à 100 %) doit être constitué par le médecin traitant, avec éventuellement l’appui du neurologue suivant le malade.
Les malades ont la possibilité d’obtenir une allocation d’adulte handicapé en déposant un dossier auprès de la Maison départementale des personnes handicapées (MDPH). Suivant leur état, une prestation de compensation du handicap peut aussi être allouée aux malades.
Cette maladie donne droit à la délivrance d’une carte d’invalidité à 80 % avec assistance d’une tierce personne, en théorie dès que le diagnostic est posé. L’orientation vers les établissements spécialisés est sous le contrôle de la Commission des droits et de l’autonomie des personnes handicapées (CDAPH), organisée au sein de la MDPH.
Pour plus de précisions, vous pouvez consulter le cahier Orphanet « Vivre avec une maladie
rare en France : aides et prestations » (ici), qui compile toutes les informations sur la
législation en cours, les aides, les modalités de scolarisation et d’insertion professionnelle
disponibles pour les personnes atteintes de maladies rares.
AMIS4- Invité
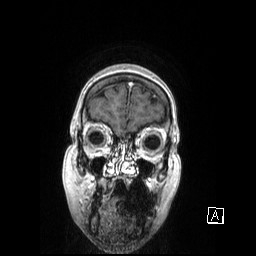
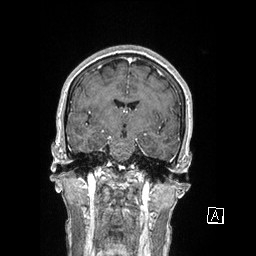
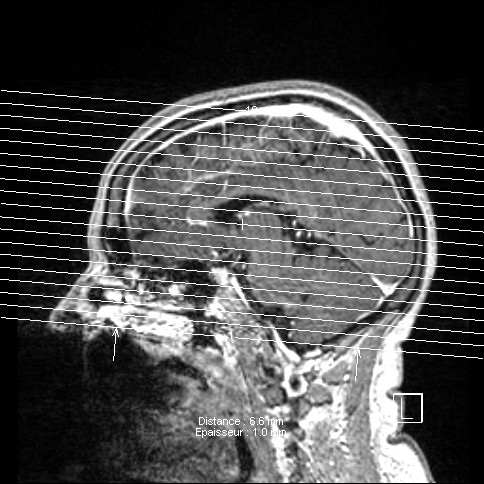
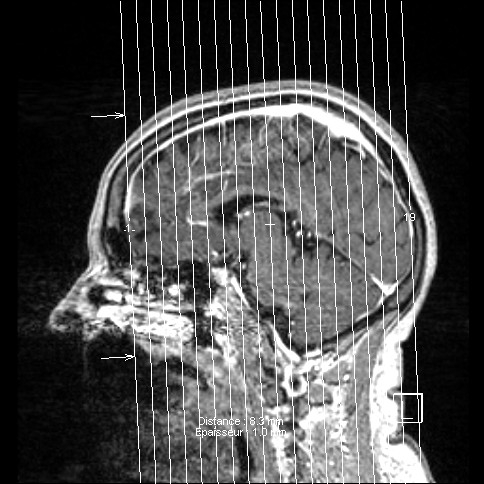
IRM SCLEROSE LATERALE AMYOTROPHIQUE FORME BULBAIRE
par estelle Dim 20 Déc - 23:43
Deux soeurs - deux parcours :
l'une chiari 1 - nombreux symptômes depuis l'enfance mais sans réponse autre que psychologique...
l'autre S.L.A. - quelques éléments indicateurs ces 10 derniers années, diagnostiquée et traitée à tort pour une pseudo thyroïde - la perte progressive de la parole, la difficulté pour se déplacer.... ont donné enfin lieu à des examens plus approfondis..
Ce qui est frappant pour les S.L.A., c'est que l'on reste lucide tout au long de la progression et jusqu'à la fin...
Alors que la pathologies Chiari semble bien affecter les capacités intellectuelles : concentration, mémoire, neurones etc...
Je posterai différentes images issues des I.R.M. que nous souhaitons diffuser pour informer d'autres familles.
Bien évidemment, seul un professionnel compétent vous aidera à comprendre et vous confirmera son diagnostic...
Une prise en charge est automatiquement mis en place dès établissement du diagnostic...
Ainsi, après des mois d'investigation auprès de médecins qui sont restés sans réponse.
L'hospitalisation et les différents examens ont enfin et malheureusement donné un diagnostic :
S.L.A. ou sclérose latérale amyotrophique (maladie de Charcot).
La progression de l'atrophie (langue, absence parole...) ont orienté rapidement sur la forme "bulbaire"...
Ce diagnostic a été posé par le CHU de STRASBOURG courant début 2008, après trois jours d'hospitalisation comportant différents examens...
N.B. Une ponction lombaire a également été réalisée -
voici les premiers que je souhaite diffuser - les clichés ne sont pas de bonne qualité et ne constitue en aucune façon
un diagnostic...
rappel d'un croquis de base de cerveau -






estelle- Admin
- Messages : 83
Date d'inscription : 20/10/2009
Localisation : AIN - BOURGOGNE - RHONE ALPES
DEMANDE INFO
par ALICE Mer 28 Avr - 10:37
cette pathologie.
Comment aider un proche atteint de S.L.A. ?
ALICE- Invité
CONTACTS
par estelle Ven 30 Avr - 6:18
Quand on a un proche atteint de la S.L.A. :
La solitude et l'impuissance sont omniprésents.
Tout au long de l'évolution, la lucidité demeure intacte pour ces patients.
Pour communiquer, nous sommes à ta disposition.
Pour avoir partagé le combat de ma sœur, je comprends tout-à-fait ce que tu exprimes.
On n'oublie jamais
Cependant, les souvenirs que vous partagerez avec ce proche.
Ce seront des moments uniques, que personne ne pourra vous prendre.
Je suis à ta disposition pour échanger, tu trouveras mes coordonnés sur la page d'accueil du site
estelle- Admin
- Messages : 83
Date d'inscription : 20/10/2009
Localisation : AIN - BOURGOGNE - RHONE ALPES

alain- Messages : 1
Date d'inscription : 27/04/2010
Age : 62
Localisation : RHONE ALPES
informations diverses à parcourir
par laura Dim 16 Mai - 8:53
http://www.sla-quebec.ca/fr/video.php?id=1
Le 21 juin est la journée nationale de la Sclérose Latérale Amyotrophique (SLA).
Ne pourriez vous pas organiser des échanges avec les canadiens, belges etc..
comparez ainsi les traitements..
le vaincre un jour ! il le faudrait :
"Amyotrophic lateral sclerosis (also known as Lou Gehrig's disease) is a devastating neurodegenerative disease, those living with the disease become progressively paralyzed due to degeneration of the upper and lower motor neurons in the brain and spinal cord. Eighty percent of people with ALS die within two to five years of diagnosis - unable to breathe or swallow. Along with ALS, neurodegenerative diseases include Alzheimer's disease, Huntington's disease and Parkinson's disease. According to the World Health Organization, neurodegenerative diseases are predicted to surpass cancer as the second leading cause of death in Canada by 2040."
A la télévision - il y a eu une émission intéressante.
Le magazine de la santé a diffusé sur France 5 - son article reprend bien les éléments généraux.
Les IRM seraient intéressantes à comparer.
Les patients conservent leur lucidité, c'est surprenant.
"La Sclérose Latérale Amyotrophique, ou SLA, est aussi connue sous le nom de maladie de Charcot, du nom du neurologue qui l'a identifié.
Il y a environ 800 nouveaux cas par an en France, surtout des personnes âgées de 50 à 75 ans, mais la SLA peut débuter plus tôt. Cette maladie neurologique se traduit surtout par des paralysies, une diminution du volume des muscles et des contractions musculaires rapides, les fasciculations (tressautements musculaires).
Ces troubles s'expliquent par une atteinte des neurones moteurs, ou motoneurones. Ce sont les cellules nerveuses qui contrôlent les muscles et qui leur commandent de se contracter.
Les motoneurones atteints dans la SLA se situent dans la moelle épinière. Mais le cerveau peut aussi être touché, au niveau du tronc cérébral, situé à la base du crâne.
C'est une des parties du tronc cérébral, le bulbe, qui peut être atteinte en premier par la SLA, d'où une altération de la parole et de l'alimentation. Mais en général, tous les muscles sont touchés, d'abord ceux des membres, pour finir par ceux de la respiration.
Les paralysies évoluent, en fonction des formes de la maladie, plus ou moins rapidement... de quelques années à dix ans, parfois plus.
De gros progrès ont été faits pour prolonger l'autonomie des malades dans de bonnes conditions. Le premier objectif est d'adapter le domicile de la personne, pour qu'elle reste chez elle en toute sécurité.
Les patients gardent toutes leurs capacités intellectuelles. C'est un facteur important pour se motiver et suivre les soins, nombreux et contraignants. Ces soins sont effectués de plus en plus tôt pour ralentir l'évolution de la maladie, en association au riluzole, cette substance ne guérit pas mais freine seulement l'évolution de la SLA. Les soins sont adaptés spécifiquement à chaque patient et aux troubles dont il souffre. C'est vraiment l'un des plus gros progrès de ces dernières années."
Autre progrès dans la prise en charge : l'association de plusieurs spécialités (neurologues, ergothérapeute, kinésithérapeute ou encore diététicienne...). Là encore, tout le travail des équipes est de s'adapter aux besoins des malades, jusque dans les stades les plus graves, quand fauteuil roulant et aides pour s'alimenter ou respirer sont nécessaires.
Il existe désormais des centres SLA dans chaque région de France qui regroupent toutes les professionnels, médicaux et paramédicaux, pour assurer aux patients une prise en charge personnalisée et globale de leur maladie.
Pour en savoir plus
* Réseau de la SLA en Ile-de-France
Site web : http://www.reseau-sla-idf.org
* Les Papillons de Charcot
Site web : http://www.lespapillonsdecharcot.com
Association.
* Association pour la Recherche sur
la Sclérose latérale amyotrophique
et autres maladies du motoneurone (ARS)
Site web : http://www.ars.asso.fr/
et cette article à parcourir :
http://sante-medecine.commentcamarche.net/contents/traitements/Sclerose-laterale-amyotrophique-has

laura- Messages : 3
Date d'inscription : 10/05/2010
Localisation : rhone alpes
message de P. BASTIAN SLA
par Admin Sam 21 Aoû - 23:12
À l'attention des membres de Sclérose Latérale Amyotrophique
Pierre Bastian 22 août, à 00:28
Bonjour à toutes et à tous ,
Cela fait très longtemps , que je ne suis pas venu vers vous et je m'en excuse !
Mais , je suis toujours au combat ! Mon groupe a changé , je ne demande plus de dons , mais votre soutien , et diffusé le message au maximum de vos contacts ! je vous donne quelques lignes de ma plaidoirie ! je vous embrasse , avant !
De vous à moi , je vais bientôt "Cr...." , alors permettez moi , de vous dire que je m'en fous , de vos effets contre productifs ! Et je m'en fous de froisser , qui que ce soit ! Mais alors royalement !
Je dénonce un très grave , problème de Santé Publique !
Je dénonce un manquement , criant de dignité !
Je dénonce une euthanasie à domicile !
Je dénonce une incompétence totale de tous les corps médicaux , dans toutes les régions , due à une méconnaissance , la plus complète de la pathologie !
Je dénonce , qu'il n'y ai aucunes structures , en France , permettant de finir nos jours , dignement , paisiblement et en toute sécurité !
Je dénonce un oubli d'accompagnement des familles , du début de la maladie , à la fin !
Leurs recherches ne sont plus productives depuis une dizaine d'années ! Tant que certains abrutis refuseront , de se diriger vers les celulles souches , on continura à perdre , du temps ! Sauf que plus les années passent , et plus il y a de patients atteints !
En 30 ans , on a trouvé le rilutek , ce n'est qu'un ralentisseur , qui agit notamment sur l'appareil respiratoire , pour prolonger un peu plus , notre agonie !!! qu'en dites vous , en 30 ans , de quoi se plaint - on !
Que voulez vous ^faire avec des mecs qui se prennent pour des dieux ???!!!
Début 80 , on a réussi à enfermer chez les fous , les cancereux et les HIV , on savait pas quoi en foutre , et le pire , on pensait qu'ils étaient contagieux ! 30 ans plus tard , on ne sait pas quoi foutre de nous , on sait qu' on est pas contagieux !
vous savez , je fais tout çà , en sachant que je serais plus la pour en profiter , mais je pourrais partir tranquille , quand je verrais que ce que j'ai amorcé , prendra forme !
Admin- Admin
- Messages : 168
Date d'inscription : 15/10/2009 -
Un relais pour la S.L.A. - un combat - un message pour la vie.
par PIERRE2 Mar 21 Sep - 9:00
à qui nous envoyons tous nos encouragements.
message du
20 septembre, à 22:00
https://www.facebook.com/?sk=messages&tid=1189165705585#!/profile.php?id=100000542919409
https://www.facebook.com/?sk=messages&tid=1189165705585
Mes Amies , mes Amis ,
Je tiens , tout d'abord à vous remercier , de tous vos témoignages qui m'ont réconforté , dans mon lit !
Mais envoyez ce message , à tous vos contacts , qui eux enverront à leurs contacts , etc ...
J'ai nommé , comme Présidente Melle BELLISARIO Amélie , qui est également , ma petite sœur de Cœur !
Elle est la Fille de mon Ami Franco ...
Je vais ensuite , nommer , comme administrateurs , plusieurs membres de cette famille , qui sont touchés par la forme la plus rare de la SLA !
C'est la forme familiale !
Cette famille est particulièrement , décimée depuis ces 30 dernières années !
Et elles sont malheureusement , très informées , par cette s. !
La 1ère qui a accepté , c'est Mme BELLISARIO Nathalie , qui est la Nièce de Franco !
Elle est également Vice-présidente de l'association SLAF !
Donc , ces personnes peuvent vous écrire , au même titre que moi !
Pourquoi cette décision ?
Parce cette famille , me coule dans le sang , et elle a toute ma confiance !
En ce qui me concerne , la maladie avance , et m'épuise un peu plus qu'avant !
Mais je continue le combat , les procédures sont en cours , sur Paris !
Je communique avec Mr le Président de la République , j'ai déjà envoyé un courrier , auquel il m'a répondu favorablement , et je prépare un second courrier !
Je gère , n'ayez crainte !
Franco , a créé une association , familiale , et il en été le Président !
Sa nièce , Nathalie Bellisario , qui est vice-présidente , m'a annoncé vouloir continuer le combat !
J'ai donc décidé , de vous en faire part , pour aider cette association , par des dons , de l'aide matérielle , etc ...
Tout est détaillé sur son site !
Je suis toujours , très affecté par sa disparition , et je communique tous les jours , avec sa fille Amélie !
Cette famille , ne sont pas des Amis ,
c'est beaucoup plus que cela , j'ai l'impression de faire partie , de leur famille !
En tous les cas , ils me coule dans le sang !
Je vous donne le lien , de son Association :
http://lesenfantsdelaslaf.free.fr/index.php?option=com_user&view=register&Itemid=19
Je dénonce un très grave , problème de Santé Publique !
Je dénonce un manquement , criant de dignité !
Je dénonce une euthanasie à domicile !
Je dénonce une incompétence totale de tous les corps médicaux , dans toutes les régions , due à une méconnaissance , la plus complète de la pathologie !
Je dénonce , qu'il n'y ait aucune structure , en France , permettant de finir nos jours , dignement , paisiblement et en toute sécurité !
Je dénonce un oubli d'accompagnement des familles , du début de la maladie , à la fin !
Leurs recherches ne sont plus productives depuis une dizaine d'années !
Tant que certains a.... refuseront , de se diriger vers les cellules souches , on continuera à perdre , du temps !
Sauf que plus les années passent , et plus il y a de patients atteints !
En 30 ans , on a trouvé le rilutek , ce n'est qu'un ralentisseur , qui agit notamment sur l'appareil respiratoire , pour prolonger un peu plus , notre agonie!!!
qu'en dites vous , en 30 ans , de quoi se plaint - on !
Que voulez vous faire avec des m. qui se prennent pour des dieux ???!!!
Début 80 , on a réussi à enfermer chez les fous , les cancéreux et les HIV ,on ne savait pas quoi en foutre , et le pire , on pensait qu'ils étaient contagieux !
30 ans plus tard , on ne sait pas quoi f. de nous , on sait pourtant, qu'on n'est pas contagieux !
vous savez , je fais tout cela , en sachant que je ne serais plus la pour en profiter , mais je pourrais partir tranquille , quand je verrais que ce que j'ai amorcé , prendra forme !
Je vous remercie infiniment et je vous embrasse ...
réponse à Pierre (publiée sur facebook et sur AMISDMOM) - solidarité des patients atteints de maladies méconnues ou orphelines.
Bonjour Pierre, j'ai parcouru avec beaucoup d'émotions votre très belle lettre.
S'il vous plait, ne renoncez jamais -
vous avez peut-être en vous, la solution.
Les cellules souches finiront par parler, s
et si vous étiez porteur de l'antidote ?
qui le sait ?
Nous avons publié votre message d'espoir parce que nous savons ce que ...c'est de se battre contre des moulins à vent.
La solidarité, l'amour sont plus forts que toutes les maladies.
n'abandonnez jamais..
Relayez - la vie c'est aussi un relais de partage, de solidarité et d'amour.
https://amis-d-mom.forums-actifs.com/echanger-partager-confronter-differentes-experiences-plusieurs-pathologies-f1/sclerose-laterale-amyotrophique-t9.htm#342
PIERRE2- Invité
LES MOUCHES - LE GLUTAMATE - SLA - les scléroses.
par MARC2 Mar 21 Sep - 11:40
Pourquoi pas, les recherches finiront bien un jour par aboutir.
Le titre du lien que je vous poste est hard - pas grave - la fin justifie les moyens.
l'article n'est pas récent.
Et il serait intéressant de chercher ou nous en sommes trois ans plus tard.
En attendant que les cellules souches parlent - parcourez - changez vous les idées avec cette étude sur les mouches, le glutamate....
les mouches ont beaucoup à nous apprendre.
BON COURAGE
La recherche sur le glutamate
Par Ophélie Neiman | Rue89 | 22/12/2007 | 21H55
"A l'occasion d'expériences en vue de guérir certaines scléroses, des chercheurs ont modifié le taux de glutamate de mouches drosophiles, etont observé avec stupeur l'apparition de parades homosexuelles parmi les insectes. Une avancée sur la relation entre comportement sexuel et communication entre neurones.
Vous pouvez observer cette hyperactivité sexuelle dans la vidéo ci-dessus,véritable séquence pornographique. Mais tout public. Là où le résultat est devenu stupéfiant aux yeux des chercheurs, c'est que ce comportement homosexuel a cessé dès que le taux de glutamate des insectes est revenu à la normale.Ce qui n'était pas le résultat attendu. Jean-François Ferveur est directeur de l'unité de recherche de Communication chimique chez les insectes à l'Université de Bourgogne,un département spécialisé dans les parades sexuelles des insectes. Il a travaillé aux côtés de Yael Grosjean, de l'université de Lausanne,ainsi sensibilisé à ces comportements :
Le même glutamate que dans la nourriture chinoise
Maisque cherchaient-ils à la base ? A bloquer la circulation du glutamate.Le glutamate est présent dans le corps humain, émis depuis les cellulesgliales et transporté autour des neurones grâce à un gène,celui-là même que les chercheurs ont bloqué sur leurs mouches drosophiles.
Or quand ce glutamate est présent en trop grande quantité, il détruit les neurones, provoque de graves maladies neuro-dégénératives,voire la mort du patient. C'est ce qui se passe, par exemple, dans le cas des scléroses latérales amyotrophiques (SLA).
Pour les amateurs de cuisine chinoise, il s'agit bien du mêmeglutamate que l'exhausteur de goût du même nom, également nocif à haute dose.
Les mouches drosophiles sont connues pour leur génome simplifié et largement utilisées pour l'étude des maladies neuro-dégénératives. Yael Grosjean adonc,grâce à des produits pharmacologiques, bloqué le gène qui permet la transmission du glutamate. Et ainsi observé le changement de omportement. <blockquote>« Quand nous stoppons la transmission du glutamate, les neurones-cibles multiplient leurs récepteurs pour tenter de capter le mieux possible la substance absente. Ils deviennent ainsi hyperactifs et vont alors détecter des phéromones mâles qui,normalement, les laissent indifférents. Mais là, ils sont hyperexcitéset les drosophiles réagissent très vigoureusement à cette odeur. »
Bientôt sur des souris
YaelGrosjean et Jean-François Ferveur s'accordent à dire que c'est le caractère temporaire, d'une durée de quelques heures, qui ouvre de nouvelles perspectives, comme l'explique ce dernier.
Selon Yael Grosjean, les conclusions sont nombreuses et loin d'être anodines :
<blockquote>« D'abord,cette expérience montre qu'un comportement, sexuel ou non, n'est pas figé et peut varier selon les transmissions chimiques entre neurones.Ensuite, le comportement vis-à-vis des femelles n'a jamais varié, alors que l'excitation envers les mâles subissait de brusques changements. Il y aurait donc deux circuits parallèles et indépendants d'excitation. »</blockquote>La découverte paraîtra sous peu dans la revue Nature Neuroscience, mais certains éléments figurent déjà sur le site internet de Nature.La relation entre le glutamate et la sexualité devrait bientôt être examinée sur des mammifères, par d'autres équipes de scientifiques. Les résultats attendus sont similaires, puisque le glutamate a les mêmes fonctions sur les insectes que sur les animaux plus évolués.
Toutefois, peu de risques de voir l'expérience transposée à échelle humaine dans les prochaines années. D'abord parce que notre génome estbien plus compliqué que celui de notre amie la drosophile.Ensuite,comme l'explique Yael Grosjean :"
« L'homosexualité n'est pas une maladie. Pas question, donc, de tenter de “guérir l'homosexualité” en triturant le taux de glutamate. »
Voici le lien à pister.
http://www.rue89.com/2007/12/22/la-recherche-sur-le-glutamate-finit-en-enculage-de-mouches
réponse cocasse
donc
- Rien n'interdirait de triturer le glutamate pour en faire un anti dote contre "l'homoSLA"
MARC2- Invité
Les cellules souches dans la sclérose latérale amyotrophique SLA - Pr Couratier et Dr Corcial P.
par Pierre Dim 13 Mar - 20:44
Objet : Les Cellules Souches dans la Sclérose Latérale Amyotrophique ...
Par le Pr Philippe Couratier, Président du Conseil Scientifique de l'ARS LA (Centre SLA de Limoges) et le Dr Philippe Corcia, Vice-Président (Centre SLA de Tours).
Les cellules souches permettent une nouvelle approche thérapeutique : réparer ou protéger les cellules nerveuses des processus responsables de la mort neuronale.
Le recours aux cellules souches dans la SLA remonte à plus de 10 ans maintenant.
Plusieurs types de cellules souches ont fait l’objet d’études dans la SLA.
Ces cellules diffèrent par leur aptitude à se différencier en cellules précurseurs ou en cellules plus évoluées ; il existe donc des cellules souches dites d’origine embryonnaire ou "pluripotentes" dans la mesure où elles peuvent évoluer vers tous les types cellulaires possibles.
Les cellules obtenues à partir du système nerveux sont dites "multipotentes" car elles peuvent se différencier uniquement en cellules nerveuses (neurones, astocytes, oligodendrocytes) qui ont pour avantage par rapport aux cellules précédentes de ne pas évoluer vers des cellules anormales malignes. Ces cellules sont prélevées à partir du cerveau, de la moëlle épinière et de l’épithélium olfactif. Une autre source de cellules souches est les cellules mésenchymateuses prélevées à partir de la moelle osseuse et enfin des cellules sanguines du cordon ombilical. Toutes ces cellules souches ne présentent pas les mêmes risques : en effet, l’utilisation de cellules embryonnaires qui peuvent se différencier en n’importe quelle cellule humaine peuvent parfois évoluer en cellules tumorales dites "tératomateuses".
Les modalités d’injection sont également multiples : directement dans le cerveau (injection parenchymateuse), dans les ventricules cérébraux qui contiennent le liquide céphalo-rachidien, par voie veineuse ou, enfin, par injection dans le péritoine qui est l’enveloppe qui
protège les organes dans l’abdomen.
Les cellules souches dans la SLA
Compte tenu du caractère diffus de l’atteinte motrice, deux attitudes prévalent dans le mode d’administration : la voie systémique (sanguine ou péritonéale) ou la voie locale (intraparenchymateuse, ventriculaire ou médullaire).
La voie systémique est envisageable car les cellules souches ont la capacité de traverser la barrière hémato-encéphalique. Cette modalité nécessite une injection d’une grande quantité de cellules et l’utilisation secondaire d’immunosuppresseurs. Les injections locales ont l’avantage de permettre le dépôt des cellules réparatrices à proximité de la région atteinte. Le statut immunologique particulier du Système Nerveux Central évite l’utilisation secondaire d’immunosuppresseurs. Par contre l’inconvénient majeur est lié au nombre de sites d’injections qui accroît le risque de lésion neurologique.
Une alternative est l’injection dans les ventricules cérébraux qui contiennent le liquide céphalo-rachidien et de permettre ainsi une diffusion tout le long du système nerveux.
Plusieurs travaux ont déjà évalué l’impact des cellules souches dans la SLA. Il s’agit dans tous les cas d’études faites sur un faible échantillon de patients différentes à la fois par le type de cellules utilisées et la voie d’administration. Ceci empêche donc toute analyse globale.
Pour illustrer l’attitude du monde scientifique sur l’utilisation des cellules souches dans le traitement de la SLA, la publication récente du travail rétrospectif faite par l’équipe hollandaise de L Van den Bergh souligne parfaitement les avancées et les interrogations qui persistent.
Nos collègues hollandais ont eu l’opportunité d’étudier avant et après l’implantation de cellules souches 7 patients SLA qui s’étaient rendus en Chine pour l’injection de cellules souches olfactives dans le tissu cérébral. Dans ce travail, l’évolution du testing musculaire,
du handicap fonctionnel par le score de l’ALSFRS-révisé et de la capacité respiratoire ont été comparés sur une période débutant de 4 mois avant l’intervention et prolongée l’année suivant l’injection de cellules souches.
Plusieurs constatations ressortent de cette étude :
1) Cette chirurgie n’est pas sans risque : en effet, 2/7 patients ont souffert de complications graves (un cas de thrombose veineuse cérébrale et un cas de décompensation respiratoire ayant nécessité une ventilation assistée) ;
2) Deux patients ont signalé une amélioration transitoire dont le caractère subjectif ne peut pas être exclu. L’éventualité d’une réelle amélioration précoce est improbable car celle-ci implique la reconstitution de connexions entre les neurones qui ne peuvent pas apparaître dans les suites immédiates ;
3) Dans la mesure où l’injection de cellules souches s’apparente à une greffe de tissus, il est surprenant de noter qu’aucun patient n’ait bénéficié d’un traitement prévenant le rejet des cellules souches et qu’aucun cas de rejet n’ait été enregistré ;
4) Chez tous les patients l’évolution de ces trois paramètres s’est toujours faite vers l’aggravation ;
5) Enfin, les auteurs restaient circonspects sur la possibilité d’obtenir à partir d’une injection focale la régénération de cellules nerveuses très à distance du site d’injection.
Conclusion :
Il est actuellement prématuré de considérer l’implantation de cellules souches comme un traitement validé de la SLA. Il n’existe pas pour le moment de preuves scientifiques indiscutables sur l’efficacité de ce type de traitement.
Les études publiées sont le plus souvent des études faites en ouvert (c'est-à-dire dans lesquelles le fait que le patient soit traité était connu à la fois du médecin et du patient). Pour des raisons évidentes, il est difficile d’un point de vue éthique de mettre en place une étude en double aveugle contre placebo. Cela est un frein majeur à la validation de ce traitement sur des effectifs de patients conséquents, tout comme le sont la variété de cellules souches et de voies d’administration utilisables qui ne permet pas de comparer les études entre elles.
https://www.facebook.com/jbhibon#!/?sk=messages&tid=1728172357166
RDV sur Facebook pour partager votre avis sur cet article.
Pierre- Invité
Listes publiées sur ORPHANET - mise à jour 2011 effective - merci
par alice2 Jeu 8 Déc - 23:17
- Cela m'a permis de comprendre l'évolution de la S.L.A. - je
comprends mieux pourquoi il est important de communiquer.
Je vous remercie pour le lien - nous nous sommes rapprochés d'un centre proche de notre région.
Le lien est mis à jour - si vous pouviez le publier
copie des centres principaux en France :
http://www.orpha.net/consor/cgi-bin/Clinics_Search_Simple.php?lng=FR&LnkId=106&Typ=Pat&CnsGen=n
- FRANCE
- ALSACE
- STRASBOURG
- CHU Hôpital Civil
- Plus d'informations
- FRANCE
- AQUITAINE
- PESSAC
- Consultation multidisciplinaire sclérose latérale amyotrophique et maladies du motoneurone - centre régional de référence
- CHU de Bordeaux - Groupe hospitalier Sud - Hôpital Haut-Lévêque
- Plus d'informations
- FRANCE
- AUVERGNE
- CLERMONT-FERRAND
- Consultation spécialisée sclérose latérale amyotrophique - centre régional de référence
- CHU de Clermont-Ferrand
- Plus d'informations
- FRANCE
- AUVERGNE
- CLERMONT-FERRAND
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHU de Clermont-Ferrand
- Plus d'informations
- FRANCE
- BASSE NORMANDIE
- CAEN
- Consultation spécialisée sclérose latérale amyotrophique - centre de compétences
- CHU de Caen - Hôpital de la Côte de Nacre
- Plus d'informations
- FRANCE
- BOURGOGNE
- DIJON
- Consultation multidisciplinaire sclérose latérale amyotrophique - centre régional de référence
- CHU de DIJON - Délégation à la recherche clinique et à l'innovation
- Plus d'informations
- FRANCE
- BOURGOGNE
- DIJON
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHU de DIJON - Délégation à la recherche clinique et à l'innovation
- Plus d'informations
- FRANCE
- CENTRE
- TOURS
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHRU de Tours - Hôpital Bretonneau
- Plus d'informations
- FRANCE
- CENTRE
- TOURS
- Consultation multidisciplinaire sclérose latérale amyotrophique - centre régional de référence
- CHRU de Tours - Hôpital Bretonneau
- Plus d'informations
- FRANCE
- HAUTE NORMANDIE
- ROUEN
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHU Hôpital Charles Nicolle
- Plus d'informations
- FRANCE
- ILE DE FRANCE
- PARIS

- Centre de référence de la sclérose latérale amyotrophique (Coordonnateur: Pr Vincent MEININGER)
- CHU Hôpital Pitié-Salpêtrière
- Plus d'informations
- FRANCE
- LANGUEDOC-ROUSSILLON
- MONTPELLIER
- Consultation spécialisée sclérose latérale amyotrophique et maladies du motoneurone de l'adulte - centre régional de référence
- CHU Montpellier - Hôpital Gui de Chauliac
- Plus d'informations
- FRANCE
- LANGUEDOC-ROUSSILLON
- MONTPELLIER
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHU Montpellier - Hôpital Gui de Chauliac
- Plus d'informations
- FRANCE
- LIMOUSIN
- LIMOGES
- Consultation spécialisée sclérose latérale amyotrophique et maladies du motoneurone - centre régional de référence
- CHU Hôpital Dupuytren
- Plus d'informations
- FRANCE
- LIMOUSIN
- LIMOGES
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHU Hôpital Dupuytren
- Plus d'informations
- FRANCE
- LORRAINE
- NANCY
- Consultation multidisciplinaire sclérose latérale amyotrophique - centre régional de référence
- CHU Hôpital central
- Plus d'informations
- FRANCE
- MIDI-PYRENEES
- TOULOUSE
- Consultation spécialisée sclérose latérale amyotrophique et maladies du motoneurone - centre régional de référence
- CHU Toulouse - Hôpital Rangueil
- Plus d'informations
- FRANCE
- NORD-PAS DE CALAIS
- LILLE
- Consultation spécialisée sclérose latérale amyotrophique - centre régional de référence
- CHRU de Lille - Centre de biologie et pathologie
- Plus d'informations
- FRANCE
- NORD-PAS DE CALAIS
- LILLE
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHRU de Lille - Hôpital Roger Salengro
- Plus d'informations
- FRANCE
- OUTRE-MER
- SAINT-PIERRE - LA REUNION
- Centre de référence des maladies neuromusculaires et neurologiques rares (Coordonnateur: Dr Claude MIGNARD)
- Groupe hospitalier Sud Réunion
- Plus d'informations
- FRANCE
- OUTRE-MER
- SAINT-PIERRE - LA REUNION
- Consultation du centre de référence des maladies neuromusculaires et neurologiques rares de La Réunion - Pôle Sud (enfants)
- Groupe hospitalier Sud Réunion
- Plus d'informations
- FRANCE
- PAYS DE LOIRE
- ANGERS
- Consultation spécialisée sclérose latérale amyotrophique - centre régional
- CHU d'Angers
- Plus d'informations
- FRANCE
- POITOU-CHARENTES
- POITIERS
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHU de Poitiers
- Plus d'informations
- FRANCE
- PROVENCE-ALPES-COTE D'AZUR
- MARSEILLE
- Centre de référence des maladies neuromusculaires et de la sclérose latérale amyotrophique (Coordonnateur: Pr Jean POUGET)
- CHU de Marseille - Hôpital de la Timone
- Plus d'informations
- FRANCE
- PROVENCE-ALPES-COTE D'AZUR
- MARSEILLE
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHU de Marseille - Hôpital de la Timone
- Plus d'informations
- FRANCE
- PROVENCE-ALPES-COTE D'AZUR
- NICE
- Centre de référence des maladies neuromusculaires et de la sclérose latérale amyotrophique (Coordonnateur: Pr Jean POUGET)
- CHU Hôpital l'Archet 1
- Plus d'informations
- FRANCE
- RHONE-ALPES
- BRON
- Consultation spécialisée sclérose latérale amyotrophique - centre régional de référence
- CHU Lyon-GH Est - Hôpital Pierre Wertheimer
- Plus d'informations
- FRANCE
- RHONE-ALPES
- BRON
- Centre de compétences des maladies neurologiques à expression motrice et cognitive
- CHU Lyon-GH Est - Hôpital Pierre Wertheimer
- Plus d'informations
- FRANCE
- RHONE-ALPES
- SAINT-ETIENNE
- Consultation sclérose latérale amyotrophique et maladies du motoneurone de l'adulte - centre régional de référence
- CHU Saint-Etienne - Hôpital Nord
- Plus d'informations
FRANCE
RHONE-ALPES
SAINT-ETIENNE
Centre de compétences des maladies neurologiques à expression motrice et cognitive
CHU de Saint-Etienne - Hôpital Bellevue
Plus d'informations
Faisons que la recherche avance - les maladies auto immunes nous concernent TOUS.
Merci
au plaisir de vous rencontrer.
Avez vous envisagé d'adhérer à Alliance Maladies Rares ? Vous seriez un plus - pour l'expérience des patients.
à suivre.
alice2- Invité
étude relayée dans la revue scientifique Neuron
par chromoso Mer 8 Fév - 21:08
Copie-collé des informations collectées.
Une collaboration internationale de chercheurs vient d’identifier une séquence répétée de l’ADN dans un gène appelé C9ORF72, chez environ un tiers des personnes touchées par la forme héréditaire de la sclérose latérale amyotrophique (SLA). Cette découverte de la cause génétique la plus fréquente identifiée à ce jour, va permettre le développement d’un simple test sanguin pour détecter le risque de SLA dans les familles à risque. Une étude relayée dans la revue scientifique Neuron.

La SLA est caractérisée par une paralysie progressive et l'espérance de vie est généralement de 2 à 3 ans à l'apparition des symptômes. C’est la troisième maladie neurodégénérative la plus fréquente dans le monde occidental. Environ 5% des cas sont familiaux.
Les chercheurs des National Institutes of Health (NIH), de l'Université de Cardiff, de l’University College London et de l'Université de Manchestersavaient qu'une région du chromosome 9 (Figure ci-dessus) est associée à la maladie, mais ont analysé les grandes sections d'ADN pour identifier la séquence particulière qui sous-tend cet effet. Ils ont prélevé des échantillons d'ADN d'une famille galloise et d’une famille hollandaise, touchées par la SLA. Pour chaque famille, les chercheurs ont étudié l’arbre généalogique détaillé avec SLA ou non. Une fois la séquence identifiée, ils ont vérifié sa présence sur un plus grand nombre d'échantillons provenant d'une cohorte finlandaise, d'Amérique du Nord, d'Allemagne et d'Italie.
La séquence répétée a été identifiée chez 46% des personnes atteintes de SLA familiale et chez 21,1% des personnes atteintes de SLA sporadique, dans la cohorte finlandaise.
Dans les cohortes européennes, un tiers des personnes atteintes présentaient des répétitions de cette séquence.
90% à 95% des personnes qui développent la SLA n'ont pas d’antécédent familial connu la recherche a donc révélé ce lien génétique chez une majorité de personnes qui avaient des antécédents familiaux de SLA, mais . Cependant, environ 21% personnes ayant la maladie mais aucun antécédent familial, présentent aussi la même mutation.
Les chercheurs estiment donc aujourd’hui qu’un test combinant la détection de cette séquence répétée d'ADN, la cause génétique la plus fréquente de SLA identifiée à ce jour, avec d'autres mutations du gène déjà connues permettrait de détecter 90% des personnes touchées par une SLA « familiale ».
Réf internet
Neuron September 21 2011 doi:10.1016/j.neuron.2011.09.010
A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. santélog
e a écrit:
La date/heure actuelle est Jeu 9 Fév 2012 - 23:13
bonjour,
Si vous souhaitez obtenir des informations sur les avancées, parcourez les revues bibliographiques du Dr Pierre-François Pradat (Centre SLA de Paris).
Je l'ai rencontré en 2008, dans des circonstances douloureuses : intégration de protocoles pour ma sœur diagnostiquée SLA forme bulbaire.
C'est un homme très chaleureux.
Si on se concentre uniquement sur le facteur héréditaire, beaucoup finirait par vivre dans la peur.
"....Dans les grandes lignes, il écrit que la physiopathologie de la SLA est multifactorielle (intervention également des cellules non motoneuronales).
Identification de la protéine TDP-43 comme
marqueur neuropathologique commun de la SLA et de certaines formes de
démence fronto-temporale.
Identification de mutations du gène codant TDP-43 chez des patients
avec une SLA sporadique ou familiale;
Rôle neuroprotecteur du lithium à confirmer..."
à suivre avec intérêt.
chromoso- Invité
degenerescence
par Nova2 Jeu 9 Fév - 14:10
A t on des nouvelles avancées ?
stopper les dégénerescences ?
merci
estelle a écrit:
bonjour,
à lire et à suivre les études du Pr Meininger Vincent et Séverine Boillée
liens
études SLA Pr MEININGER Vincent et Séverine BOILLEE
Ou sur moteur de recherche
http://grands-prix-2011.institut-de-france.fr/communiques/nrj.pdf
http://polemsn.fr/base-documentaire/sclerose-laterale-amyotrophique/
La date/heure actuelle est Jeu 6 Sep 2012 - 12:17
Message à publier S.V.P.
N.B. à vérifier - Vérification non effectuée ce jour, prévu prochainement.
mail reçu
"Madame,
Monsieur,
ICARE
est une agence spécialisée dans les études médicales et pharmaceutiques et nous réalisons actuellement une étude auprés de patients et d'aidants sur la Maladie De Charcot / Sclérose latérale amyotrophique.
Nous recherchons pour cela [b]des patients diagnostiqués « Maladie De Charcot / Sclérose latérale amyotrophique » ainsi que des aidants de patients atteints de cette maladie.
Cetteétude sera réalisée sous forme d’entretien individuel de 60 minutes (téléphonique ou face face) et aura lieu entre le 20 septembre 2012 et le 11 octobre 2012.
Cet entretien sera mené conformément aux codes de conduite régissant les études de marchés ; l'identité du patient demeurera confidentielle tout au long de l’étude. L’entretien sera enregistré sur support audio à des fins de normes qualité et analyse uniquement. Tout ce dont le patient discutera avec nous sera traité dans la plus stricte confidentialité et conformément aux codes de conduites EphMRA, ESOMAR, PBIRG, BHBIA, MRS régissant les études de marchés.
La participation à cet entretien est volontaire, et le patient/aidant peut l’interrompre à tout moment. Il peut aussi refuser de répondre à des questions si il le souhaite.
Bien entendu, cet entretien n'a aucune vocation de prospection commerciale, seule leur opinion nous intéresse. Nous garantissons la plus parfaite confidentialité aux personnes participant à nos études.
Chaque participant recevra une indemnisation de 50 euros, qu’il pourra conserver ou reverser à l’association caritative de son choix. Pour vous remercier de votre participation, vous recevrez une indemnisation de 75 euros par patient recommandé et participant à l'entretien ; indemnisation que vous pourrez conserver ou reverser à l’association caritative de votre choix.
Si vous souhaitez nous aider dans notre recherche patient, veuillez contacter mes collègues Madame Sylvie Breux au 03 20 05 83 39 ou Madame Sandrine DAEL au 03 20.05 83 41.
Veuillez recevoir, Madame, Monsieur, l'assurance de notre considération distinguée."
Cécile Verdure International
Fieldwork Coordinator - ICARE
Direct line: +33 3 20 05 82 73
Email: [b]cecile.verdure@icare-mr.com
Nova2- Invité
MACON 71 Saône et Loire - la ville et un collectif d'accueil se mobilise pour Gilles et faire connaitre la S.L.A.
par Admin Mer 25 Mai - 11:53
 L’ association a pour but :
L’ association a pour but :– d’aider les malades atteints des maladies orphelines ou méconnues telles que :
syringomyélie – malformation ou syndrome de A. Chiari – fibromyalgie – S.F.C. syndrome de fatigue chronique – myelinolyse – compression médullaire- Scléroses en plaques – S.L.A. : sclérose latérale amyotrophique/maladie de Charcot – Autisme – et autres.
Sa présidente Mme Estelle Lussiana, a accompagné sa sœur atteinte de la maladie de Charcot
- Groupement de personnes bénévoles et solidaires
« Association constituée dans le but d’apporter un soutien et une aide à la personne en situation de handicap, aux parents d’enfants malades ou familles/patients qui se trouvent isolés, mais également confrontés à l’errance d’un diagnostic, au désarroi dans le parcours parfois, chaotique des maladies rares, orphelines et/ou méconnues.
Nous essayons d’apporter un relais en aiguillant raisonnablement, en mettant en relation des personnes handicapées et touchées par une même pathologie, en soutenant dans la limite de nos moyens, des associations ou des actions spécifiques.
Nous essayons de répondre à vos attentes, mais aussi de vous donner des pistes à explorer, nous assistons ou organisons des conférences, nous essayons de vous tendre une main de vous donner un sourire, de vous transmettre un geste de SOLIDARITÉ…. »
Siège social Gennevilliers
L'association AMIS d MOM soutient le combat de plusieurs patients. Elle vous invite à rejoindre le mouvement de Gilles. Un grand merci pour votre participation.
LA LONGUE ROUTE DES MALADES DE LA S.LA fait étape à Mâcon - Saône et Loire
le vendredi 3 juin 2016 Rdv 17 h 30 Esplanade Lamartine (face à l'Hôtel de Ville).
Ce sera la 5ème étape pour Gilles Houbart et ses amis qui engagent un défi sous forme de relais,
de la Savoie à l’Hôpital de la Pitié Salpêtrière à Paris, 788 km, 11 étapes qu’ils vont franchir du 30 mai au 11 juin en fauteuils motorisés, en parapente, à cheval, ou en joëlette...
pour expliquer que la S.L.A. (Sclérose Latérale Amyotrophique ou maladie de Charcot) est une maladie qu’il faut sortir de l’anonymat et qu’il faut vaincre par tous les moyens.
Dans le cadre du Printemps du Handicap - AMI 71,
nous avons le plaisir d'accueillir : LA LONGUE ROUTE DE LA S.L.A.
le vendredi 3 juin à l’esplanade Lamartine à Mâcon dès 17 h 30.
Nous choisissons de renforcer l’espoir, susciter des énergies nouvelles, vivre des moments inoubliables.
Ensemble nous allons partager un projet extraordinaire et grandir avec lui.
N’hésitez pas, venez avec une envie d’aller à la rencontre des relayeurs en marchant, ou en roulant, venez avec un instrument de musique, une chanson, un petit mot à offrir, une danse, un témoignage, venez avec vos voisins et vos amis...Le collectif d’accueil, soutenu par la municipalité, s’organise pour proposer des jeux, de la musique, des informations et le verre de l’amitié.
Participation non exhaustive :
Sportifs, pom pom girls : Out laws cheers de Mâcon, accordéoniste Mr Vacheresse, participation des cornemuses "les sonneurs de Killimor3, escorte Harley Davidson.....
Pour démarrer ces festivités et cet accueil , le Cinémarivaux projettera le film : "DEBOUT"
ou l'on retrouvera la talentueuse et généreuse Clémentine Célarié.
Elle oeuvre avec dévouement, au profit de la recherche sur la S.L.A.
La projection est programmée : le lundi 30 mai à 20 h, elle sera suivi d’un débat animé par Patrick Halet et Nadine Gonin sur la maladie de Charcot en préambule à l’arrivée de Gilles Houbart, prévue le vendredi 3 juin 2016 à l'Esplanade Lamartine.
En savoir plus ?
http://www.longueroutesla.org/
C’est parti, diffusez sans relâche.
Itinéraires :
Voici un lien - partie Neuville/Saône à Mâcon
https://goo.gl/maps/t173jp8T2fM2
Pour tous compléments d'informations : N'hésitez pas à joindre
[size=16]Luc Martinon [/size]
[size=16]Chargé de communication
La longue route des malades de la SLA
06 17 46 75 51
www.longueroutesla.org
Du 30 mai au 11 juin, 788 km en fauteuil roulant électrique contre la SLA
[/size]
Dernière édition par Admin le Ven 19 Jan - 13:39, édité 1 fois
Admin- Admin
- Messages : 168
Date d'inscription : 15/10/2009 -
DIFFUSION DU FILM DEBOUT de Clémentine Célarié et
par CINE MACON 05 2016 Jeu 26 Mai - 20:27
Rdv lundi 30 mai 2016 au cinémarivaux à Mâcon
diffusion du film Debout à 20 h suivi d'un débat.
Faites suivre
LIENS FB
https://www.facebook.com/photo.php?fbid=10207930450692418&set=a.3834219947282.147251.1631442287&type=3&theater
CINE MACON 05 2016- Invité
DECOUVERTE
par AMIS ESPOIR Ven 19 Jan - 13:13
voici un lien intéressant
http://www.techno-science.net/?onglet=news&news=16989
Le mécanisme moléculaire responsable du développement de la maladie de Charcot dévoilé
maladie de Charcot cerveau

Figure: Dans les motoneurones des patients atteints de la maladie de Charcot, le ribosome traduit les séquences répétées du gène C9orf72 dans les trois cadres de lecture pour produire des polydipeptides neuro-toxiques.
 Franck Martin
Franck MartinLa maladie de Charcot, également appelée Sclérose Latérale Amyotrophique (SLA), a récemment été mise sur le devant de la scène médiatique française par le décès au cours de l'été 2017 de l'ex-tennisman professionnel Jérôme Golmard. Cette pathologie, généralement fatale et incurable, est caractérisée par la neuro-dégénérescence des motoneurones entrainant une paralysie progressive des muscles. Le désormais célèbre « Ice Bucket Challenge » (défi du seau d'eau glacée), dont la pratique a été largement relayée par les réseaux sociaux, a permis de récolter des fonds dédiés à la recherche sur la SLA. Grâce à ce défi, d'importants moyens financiers ont été mis à la disposition des chercheurs pour leurs travaux sur cette pathologie. Cependant, malgré de nombreuses études, les mécanismes moléculaires mis en jeu au cours du développement de cette maladie demeuraient inconnus.
Les chercheurs ont percé ce mystère en décortiquant pour la première fois le mécanisme moléculaire responsable du développement de la maladie de Charcot. Dans toute cellule, les protéines sont synthétisées par une machinerie macromoléculaire complexe appelée ribosome. Les chercheurs ont montré que des séquences, répétées anormalement un grand nombre de fois dans le gène C9orf72 chez les patients, attirent de manière aberrante le ribosome qui se met alors à traduire ces séquences répétitives en polydipeptides. Ces études ont permis en particulier de localiser précisément le site de démarrage de la traduction et également de démontrer qu'un décalage du cadre de lecture (« frameshift ») est nécessaire au cours de la synthèse des polydipeptides par le ribosome.
Ces polydipeptides sont hautement toxiques pour la cellule parce que leur accumulation dans les motoneurones entraine leur agrégation qui provoque la mort cellulaire. Ces travaux permettent de mieux comprendre les mécanismes moléculaires qui aboutissent à la synthèse et l'accumulation de ces polydipeptides dans les motoneurones des patients. Ils ouvrent de nouvelles perspectives de traitements thérapeutiques visant à bloquer cette synthèse protéique aberrante.
Il est important de noter que d'autres maladies neuro-dégénératives (Ataxie, Maladie de Huntington, syndrome du l'X fragile etc...), également liées à l'apparition de séquences répétées, semblent suivre les mêmes règles moléculaires. De ce fait, cette découverte constitue une étape majeure pour l'amélioration de la santé humaine dans la lutte contre ces maladies.
Franck Martin
AMIS ESPOIR- Invité
Re: Sclérose latérale amyotrophique
par Admin Mar 10 Juil - 7:41
Reconnu dans le monde entier pour ses recherches sur les trous noirs et ses ouvrages de vulgarisation scientifique, Stephen Hawking, atteint de la maladie de Charcot.
"Interrogé par le BMJ s'il savait pourquoi son état avait évolué différemment d'un cas typique de MND, le professeur Hawking a répondu: "Je crois que la maladie du motoneurone est un syndrome qui peut avoir différentes causes. Peut-être que ma variété est due à une mauvaise absorption des vitamines. "
Le professeur Hawking complète son alimentation avec des comprimés minéraux et vitaminiques quotidiens, et le zinc, les capsules d'huile de foie de morue, l'acide folique, le complexe de vitamine B, la vitamine B12, la vitamine C et la vitamine E seraient particulièrement utiles. Il suit également un régime sans gluten et huile végétale et évite les aliments de commodité; tout récemment, il a commencé à inclure une petite quantité de produits laitiers https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1123440/"
Admin- Admin
- Messages : 168
Date d'inscription : 15/10/2009 -
Re: Sclérose latérale amyotrophique
par Admin Mar 10 Juil - 7:59


Admin- Admin
- Messages : 168
Date d'inscription : 15/10/2009 -
A.M.I.S. des M.O.M :: OBJECTIFS ASSOCIATION DE PATIENTS SOLIDAIRES/ OBJECTIVES OF PATIENT SOLIDARITY ASSOCIATION :: Cliquer ici- pour accéder aux différents sujets-FORUMS-DIVERS - diffusions LIENS - AMIS D MOM - Click here to reach the various subjects - FORUMS-
» mutuelle complémentaire obligatoire en 2016
» Quelles actions concrètes pour notre association de patients cette année ?
» spina bifida - méningocèle - Myéloméningocèle
» Mon histoire avec la Malformation de CHIARI de type 1
» CHIARI - REEDUCATION APRES UNE OPERATION- HELP
» Une association de patients solidaires, internationales, réseaux sociaux/Patient organization supportive, international, social networks
» la maladie de Paget
» Des conseils pour gérer une maladie méconnue ?